Zellweger UK explains how the charity is working to support research into Zellweger spectrum disorders.
The Zellweger Spectrum Disorders (ZSD) comprise a group of genetic disorders which are named after Prof. Hans Ulrich Zellweger (1909–1990), a Swiss-American pediatrician, and are characterised by the loss of functional peroxisomes. Peroxisomes are membrane-bound subcellular organelles, which compartmentalise enzymes involved in several crucial metabolic processes such as the breakdown of fatty acids, the production of ether lipids (e.g., myelin sheath lipids) and bile acids, the metabolism of hydrogen peroxide (H2O2) and the maintenance of cellular redox balance. They are present in all cells in the human body, and indispensable for human health and development.
Molecular basis and disease pathophysiology
Much progress in understanding the molecular defects and pathophysiology of peroxisomal disorders has been made by studying peroxisome function and biogenesis (i.e. assembly) in different cell models, through research with knock-out mouse models. Peroxisomal disorders are grouped into single enzyme deficiencies, including the most common defect X-linked adrenoleukodystrophy (X-ALD) and the peroxisome biogenesis disorders, in which the functional assembly of the organelle is affected1.
The single enzyme deficiencies are usually characterised by a defect in one specific peroxisomal function or metabolic pathway. However, when the affected protein is involved in the biogenesis and maintenance of peroxisomes, multiple or all peroxisomal functions can be affected. Those peroxisome biogenesis proteins are named peroxins, which are encoded by PEX genes. Up to now, mutations in 13 different PEX genes have been identified to cause a ZSD. Most peroxins function in the formation of the peroxisomal membrane or in the import of peroxisomal enzymes into the organelle. Mutations in peroxins can therefore either result in the complete absence of peroxisomes, due to a defect in their formation, or in empty membrane compartments (‘ghosts’), due to defects in the import machinery.
When peroxisomal enzymes are not imported into the peroxisomes, they cannot function or are degraded. This has major consequences for most of the metabolic pathways located in peroxisomes. A loss of peroxisomal functions is generally accompanied by the accumulation of substrates which can only be processed in peroxisomes and may be toxic (e.g. very-long chain fatty acids (VLCFAs), phytanic acid, a branched-chain fatty acid from dairy products, or bile acid precursors), and a shortage of peroxisomal products (e.g. myelin sheath lipids and bile acids).
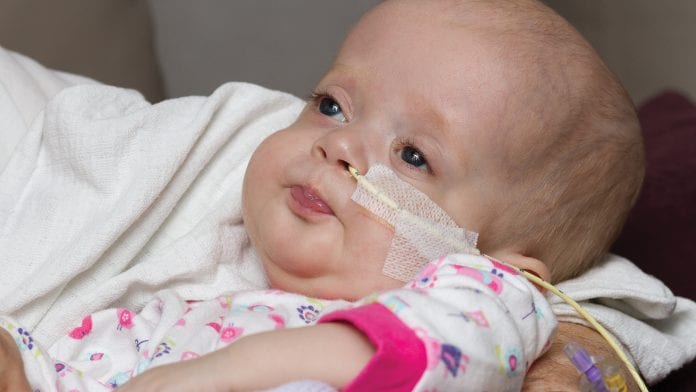
Peroxisomes are required for normal brain development and function and the formation of myelin, which insulates neuronal cells. As functional peroxisomes are important during development, patients suffering from Zellweger Spectrum Disorders already show symptoms from birth such as craniofacial dysmorphy, hypotonia, neuronal myelination and migration defects, failure to thrive and liver dysfunction. Many children fail to pass their newborn hearing screening. As the disease progresses children commonly deal with global developmental delays, vision impairment, multi-sensory loss, adrenal insufficiency and seizures. Children may also suffer other common symptoms such as renal stones, low bone density and blood clotting issues that can lead to hemorrhage and intracranial bleeding.
Even as a group, Zellweger Spectrum Disorders are rare diseases that affect an estimated 1/50,000 individuals. Severe PEX gene mutations are often fatal, and affected children do not live into adulthood while dealing with global developmental delays, deaf-blindness/multi-sensory loss and a plethora of other health issues. However, the spectrum is wide with a varying degree of disease severity, and life expectancy ranges from days old for severely affected patients to patients with milder defects who live into their 30s or even beyond.
Diagnosis and Therapy
As Zellweger Spectrum Disorders are progressive disorders, early diagnosis is important2. Laboratory diagnosis is often performed in specialized metabolic laboratories. However, the development of cost-effective next generation sequencing technologies have increased the early identification of patients through DNA testing. Once their PEX gene mutation have been identified, further children can be diagnosed by genetic testing either in utero or after birth.
It is also possible to determine whether healthy siblings and other members of the family are carriers once each parent’s mutations is identified. Laboratory diagnosis involves blood and urine analysis, and the level of plasma VLCFAs is usually an informative initial screen. Additionally, analysis of ether lipids in erythrocytes, and phytanic acid levels is performed. Those tests are preferably followed by detailed biochemical and morphological studies in patient skin fibroblasts.
Abnormalities in ZSD patients already develop in utero, and postnatal treatment options are limited. Currently there are no effective treatments for ZSD. Therapies are mostly supportive, aiming to improve the developmental outcome, survival and quality of life such as cortisol supplementation for adrenal insufficiency, anti-seizure medications, vitamin K for blood clotting disorders and pamidronate infusions for children who suffer with low bone density.
However, strategies are being developed to correct the different biochemical abnormalities in patients with milder phenotypes and less pronounced abnormalities, for example by the reduction of accumulated substrates (dietary regimens to reduce VLCFAs and phytanic acid) or replacing deficient peroxisomal products (e.g. supplementation of docosahexaenoic acid).
Furthermore, drugs are investigated for their potential to increase the expression of peroxisomal genes which can either complement the function of the disease gene or increase the number and enzyme content of peroxisomes. There are also approaches underway to identify new compounds which stabilise mutated peroxins to prevent their degradation and recover or improve protein import.
Supporting children and families
Life with ZSD can feel incredibly lonely. Most doctors and paediatricians have never even heard of peroxisomal disorders, so specialists experienced in the condition are very few and far between. Zellweger UK is a small, entirely parent-run charity for families impacted by ZSD.
Set up in 2016, the relatively new charity is run on a voluntary only basis, with the team dedicating as much of their time as possible to Zellweger UK; some of them while parenting children with extremely complex care needs, others while coping with the death of their child. The charity has no chief executive or paid employees and all funds raised go directly to supporting both research and the families affected by this devastating disorder.
Aims are to offer emotional support and to sponsor families, e.g. towards non-state funded equipment, medical aid or travel to specialist centres. The team is driven by their experiences with their own affected children and are proud to provide support for 52 families across the UK and Ireland. It can be very isolating to be told that your child’s condition is so rare that there are no other cases in your area; it is therefore a comfort to know that you have somewhere to turn and where you can connect with the only other people who can possibly truly understand what you are going through.
One of the biggest successes of the charity are the annual family weekends, which bring families on the Zellweger spectrum together, and also face to face with metabolic specialists and scientists. The families greatly enjoy putting faces to the research on peroxisomes and are eager to expand the working relationships that they are building with physicians, scientists and other international charities and patient organisations.
The biggest challenge as a rare disease charity is still the frequent societal assumption that funds only help a tiny minority; it can be difficult to inspire people to support a charity for a condition they have never heard of. A child suffering with a condition as rare as ZSD needs a cure every bit as much as a child with a more common condition.
However, the charity has amazing support from their families and a large part of the funding comes from fundraisers by families or friends. Even after they have lost their battle to this devastating disease, the children continue to inspire people and many fundraisers are held in their names.
A crucial aim of the charity is therefore to raise awareness of this devastating disease and to grow enough to be able to better support research into desperately needed, effective treatments. Peroxisomal disorders are rare and therefore receive little funding towards research. However, recently important novel functions of peroxisomes as signalling platforms, in anti-viral defence, combat of pathogens, protection against cellular stress and in the modulation of cellular redox balance have been identified which link them to aging and the development of chronic diseases such as neurodegeneration, diabetes, and cancer.
There is the hope that these new discoveries will impact on research funding, foster interdisciplinary links between industry, life and medical sciences, and develop novel leads for drug discovery and therapies for a growing list of serious human diseases in which peroxisomes are involved.
References
- Waterham HR, Ferdinandusse S, Wanders RJ (2016) Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta 1863:922-33.
- Braverman NE, Raymond GV, Rizzo WB, et al. (2016) Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab 117:313-21.
Co authors
Professor Michael Schrader, Biosciences, University of Exeter, UK
Professor Hans Waterham, AMC, Amsterdam, The Netherlands
Kerry Hughes
Treasurer
Natasha Anderson-Hunt
Chair
Carly Burton
Vice Chair
Zellweger UK
+44 (0) 1460 68405
Admin@zellweger.org.uk
Tweet @ZellwegerUk
http://www.zellweger.org.uk/
This article will appear in issue 7 of Health Europa Quarterly, which will be published in November 2018.

















