
Professor Dr François Eyskens of PCMA vzw discusses early diagnosis of MPS diseases by creating awareness and using a tandem mass spectrometry screening method.
MPS diseases, or mucopolysaccharidoses (lysomal storage disorders), are multisystem disorders that show a broad clinical spectrum and a slow to fast clinical progression. MPS diseases are autosomal recessive or X-linked recessive (MPS II) inherited disorders caused by a deficiency of a specific lysosomal enzyme, characterised by the accumulation of incompletely degraded glycosaminoglycans (GAGs) in connective tissue cells, leading to widespread organ dysfunction.
Birth prevalence of all LSDs is estimated to be around 1:10,000 life births (approximately 15% of diagnoses in the field of metabolic diseases); the prevalence of individual MPS diseases is between 1:100,000 – 1:170,000, so that they fulfill the criteria of rare disorders; the combined prevalence of MPS diseases is estimated around 1:22,000 (about 35% of all LSDs).
The disease spectrum is broad and while most physicians are not aware of the existence of these rare diseases, the diagnosis is significantly delayed in most patients. Especially in the more attenuated or milder subtypes (delay of proper diagnosis between 4-54 years after appearance of first clinical symptoms and signs), misdiagnosis is common.
The prevalence of known patients in Flanders is less than 50% of the presumptive number based on and compared to the situation in The Netherlands. Since effective disease-specific treatments, including enzyme replacement therapy and stem cell transplantation, have become available and timely initiation of these treatments is necessary to prevent the development of severe, disabling and irreversible manifestations, early diagnosis has become essential (McGill JJ, Inwood AC, Coman DJ et al. Enzyme replacement therapy for MPS VI from 8 weeks of age-a sibling control study. Clin Genet 2009; doi: 10.111/.j.1399-0004.2009.01324x).
As musculoskeletal manifestations are common, raising awareness for better recognition of these metabolic diseases based on the presenting signs and symptoms by all doctors who may encounter these patients is mandatory: paediatricians, orthopaedic surgeons, rheumatologists, child neurologists and neurosurgeons.
When the diagnosis of MPS diseases is suspected, a blood filter paper is filled with whole blood dried under ambient temperature conditions and sent to UZA – PCMA for analysis by LC-MS/MS, which is performed in a multiplex manner so that different MPS diseases can be screened for on the same blood spot at the same time. The diagnosis is confirmed by demonstration of a specific enzyme deficiency, usually in peripheral blood leucocytes and molecular genetic analysis.
New methods have been developed that quantitate lysosomal enzyme activity in dried blood spots (DBS). Enzyme activity determination as a method for screening is based on the observation by Chamoles and co-workers that lysosomal enzymes retain latent activity in DBS and can be assayed after rehydration in a suitable buffer (Gelb et al., J Inherit Metab Dis, 2006).
Starting in April 2013 we optimised, validated and evaluated a 4-plex enzymatic activity LC-MS/MS screening method (based on the method by Spacil et al., Clin Chem, 2013) for the treatable LSDs Gaucher’s disease, Fabry’s disease, Pompe’s disease and MPS I at the PCMA vzw laboratory. We proved that this method is accurate, fast, is low cost and easy to implement and therefore suitable for LSD diagnostics and selective screening. For the screening of the treatable MPS I (Hurler-Sheie), MPS II (Hunter), MPS IVA (Morquio A) and MPS VI (Maroteaux-Lamy), new and improved reagents are available (Gelb et al., Anal Chem, 2014) and the method was validated in the PCMA lab.
Cost effectiveness
In most of the rare disease cases, an earlier detection and start of treatment leads to lower costs and a better quality of life, compared to those who are diagnosed in a late disease stage. As stated in Fig. 1, rare disease treatment costs account for a comparatively high percentage within national healthcare total expenditures; these circumstances make cost saving policies for rare disease treatment a high priority, as presented in the Belgian plan for rare diseases.
Value creation framework
Enhancing the diagnostic capability of mucopolysaccharidoses (MPS diseases) will carry out positive effects on a diverse set of stakeholders, each of them possessing different degrees of engagement. The following value creation framework is built (Fig. 2).
Value for patients
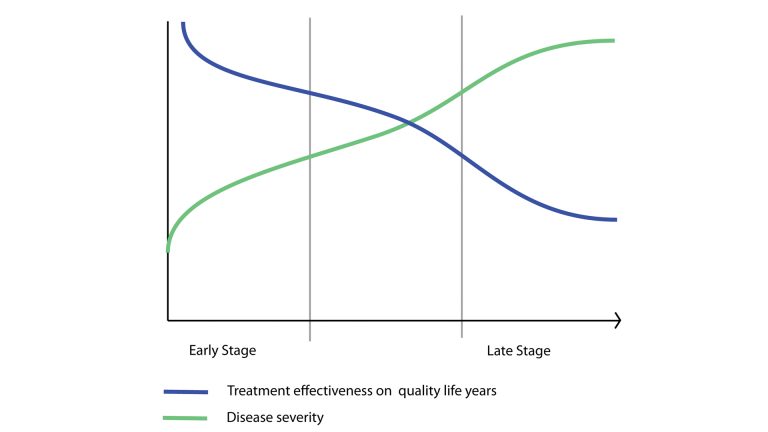
From a social and ethical perspective, rare disease affected patients are beyond question the main beneficiaries of this initiative. Most patients suffer a disappointing medical experience in respect to their quest for a rare disease diagnosis. A variety of weak signals, tiny clinical or test details go unnoticed and underestimated, resulting in life-threatening late diagnoses. The positive side effects for earlier diagnosis are virtually limitless. Most of the time an early diagnosis means the difference between a future normal life or the condemnation to a lifelong condition as physically and mentally disabled. It means the difference between life or death. Most of the time, being detected earlier and with greater accuracy means receiving the most appropriate treatment and continued opportunity of life. Even if a treatment doesn’t exist yet early diagnosis can give the hope of taking part in clinical trials for drugs still in the research stage. Fig. 3 below is a conceptual chart showing how the efficacy of the treatment intended, as its power to increase the quality and length of life of patients, is inversely related to time of the diagnosis.
From an economic perspective, a patient’s interest in receiving early diagnosis translates into the possibility to afford the treatment and avoid expenses for unproductive tests and consultations. Usually, most of the expenses are covered by national insurance policies, but part of them may still be charged to the patient.
Value for the scientific community
The Antwerp University Hospital is a tertiary referral centre and rare disease reference centre in Belgium and part of the international scientific community. Patients with rare diseases are diagnosed and treated in a multidisciplinary approach.
The knowledge will be shared through publications, lectures at national and international meetings and the organisation of postgraduate courses and training. In addition, increased efficiency in rare disease-related research may lead to scientific discoveries that may apply to a broader range of drugs and medical treatments.
Value for the government and society
Providing affordable and quality healthcare for citizens is at the core of any public welfare policy. Contributing to the resolution of a relevant social and ethical issue, such as a rare disease patient’s support, constitutes a big leap along the path of ever increasing healthcare quality. Rare disease treatment costs account for a comparatively high percentage within national healthcare total expenditures. These circumstances make cost-saving policies for rare disease treatment a high priority. In regard to improving early diagnosis of Lysosomal storage diseases, MPS diseases can play a major role in saving costs for any social community and granting a better quality of life to patients.
Value for the pharmaceutical industry
Early and more accurate diagnosis can cause positive indirect effects for the pharmaceutical industry by improving patient recruitment, treatment monitoring and efficiency. Pharmaceutical companies operating in the orphan drugs sector face considerable difficulty in developing new products for rare diseases; as the very name suggests, drugs for rare diseases are seldom searched for because of the limited market attractiveness, poor and scattered knowledge, as well as trial design complexity.
Read more in Professor Eyskens’ booklet, Diagnosing Mucopolysaccharidoses diseases in young people


















